
Theoretical Exploration of Chemical Structures and Reactions
Precursors of the Charge-Transfer-to-Solvent States in I- (H2O)n Clusters
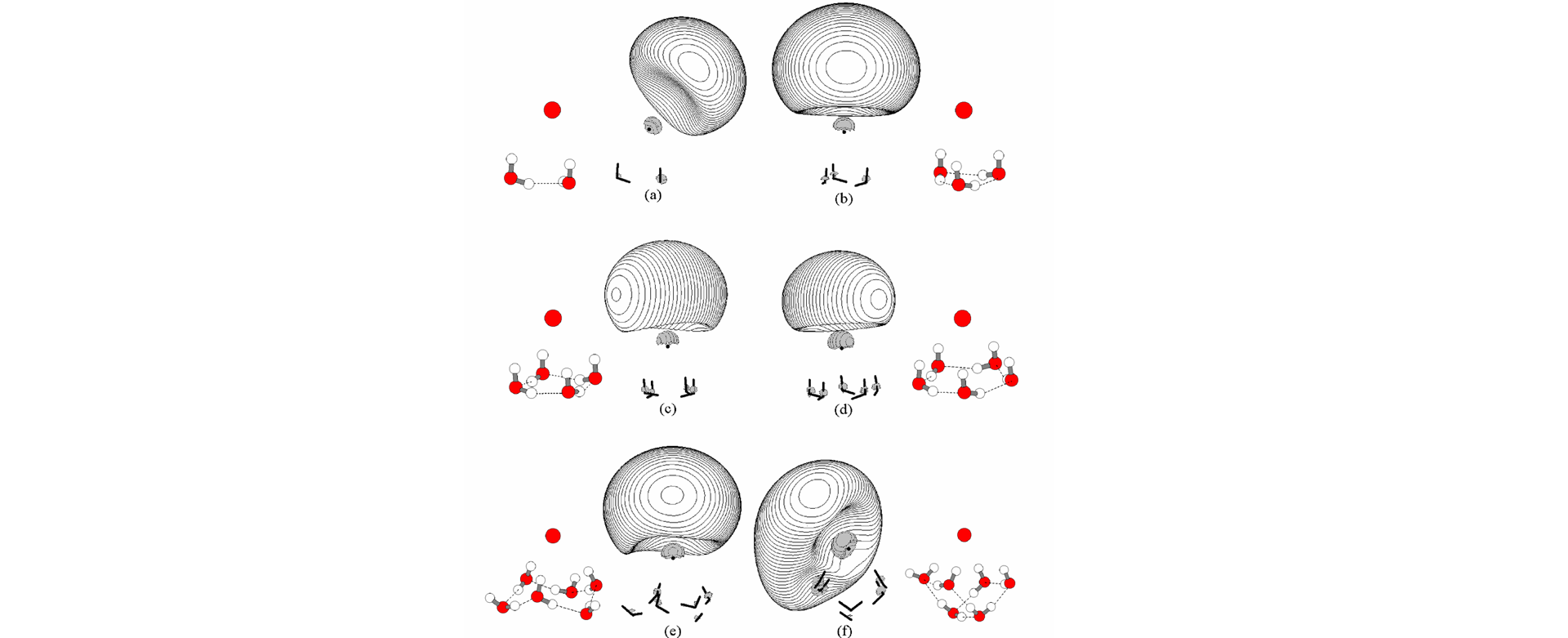
An ab-initio theoretical study of the precursors of the charge-transfer-to-solvent (CTTS) states in I-(H2O)n clusters is presented. While there is no bound excited state in monohydrated iodide I- (H2O), the CTTS precursor states, denoted as I-(H2O)n*, emerge at cluster size n³2, which confirms a recent experimental observation [Serxner et al. J. Chem. Phys. 1996, 105, 7231.]. In addition, two or more bound excited states are found for larger clusters. The absorption maximum of the interior structure of I-(H2O)6 is found to be 5.02 eV, comparable to the experimental value of 5.48 eV found in the bulk, indicating that the first hydration shell of the aqueous halide makes a very significant contribution to the solvation energy of the lowest CTTS state and that the molecular details of solvent molecules play an important role in forming the CTTS states. Comparing the CTTS precursor states I-(H2O)n* with the electronic states of the corresponding water cluster anions, e-(H2O)n, shows that the excited electron distributions are excluded from the region occupied by the electrons of the iodine atom, which in turn results in higher energies for I-(H2O)n* compared with e-(H2O)n. Moreover, it is shown that the cluster size dependence and isomer specificity of the excitation energies and absorption intensities of the I-(H2O)n clusters may provide a diagnostic tool in determining the predominate structure, surface or interior, of I-(H2O)6.
Characterization of Excess Electrons in Water Cluster Anions via Quantum Simulation
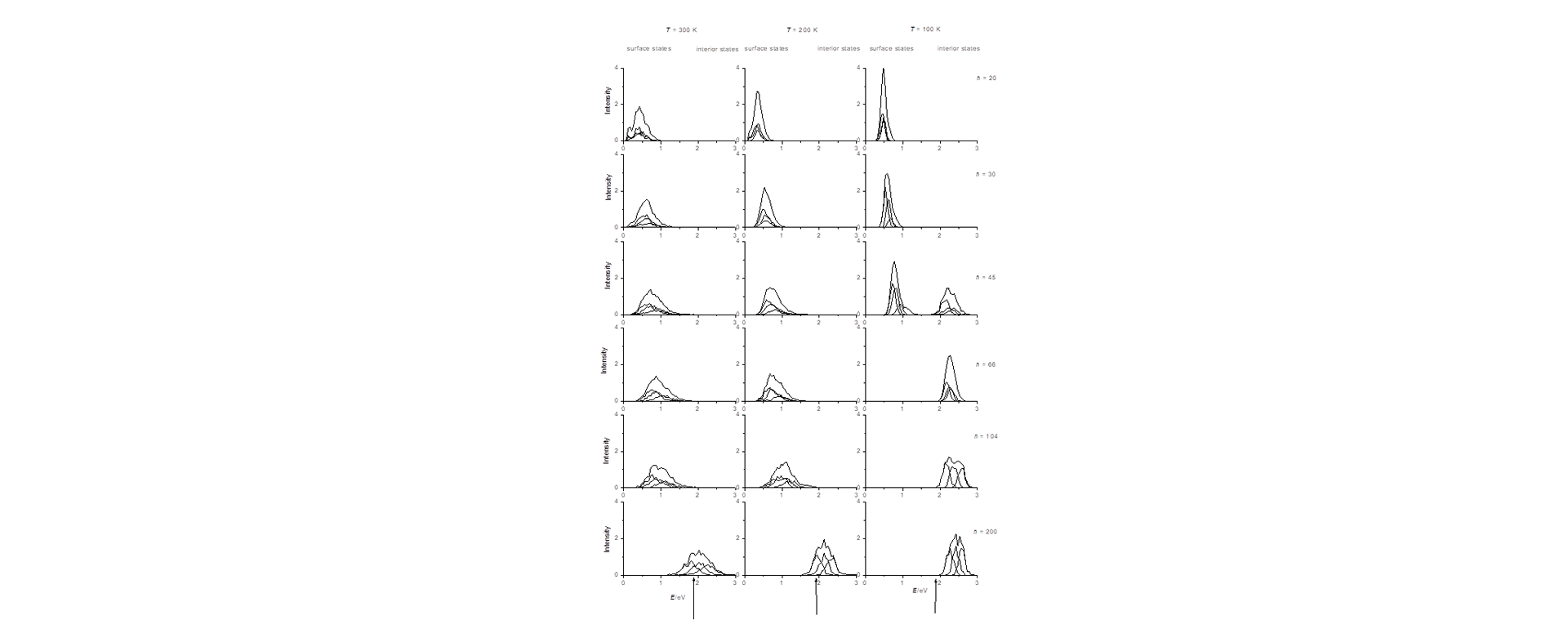
Water cluster anions can serve as a bridge to understand the transition from gaseous species to the bulk hydrated electron. However, debate continues regarding how the excess electron is bound in (H2O)n-, as an interior, bulk-like, or surface electronic state. To address the uncertainty, the properties of (H2O)n- clusters with 20 to 200 water molecules have been evaluated by mixed quantum-classical simulations. The theory reproduces every observed energetic, spectral, and structural trend with n that is seen in experimental photoelectron and optical absorption spectra. More importantly, surface states and interior states each manifest a unique signature in the simulation data. The results strongly support assignment of surface bound electronic states to the water cluster anions in published experimental studies thus far.
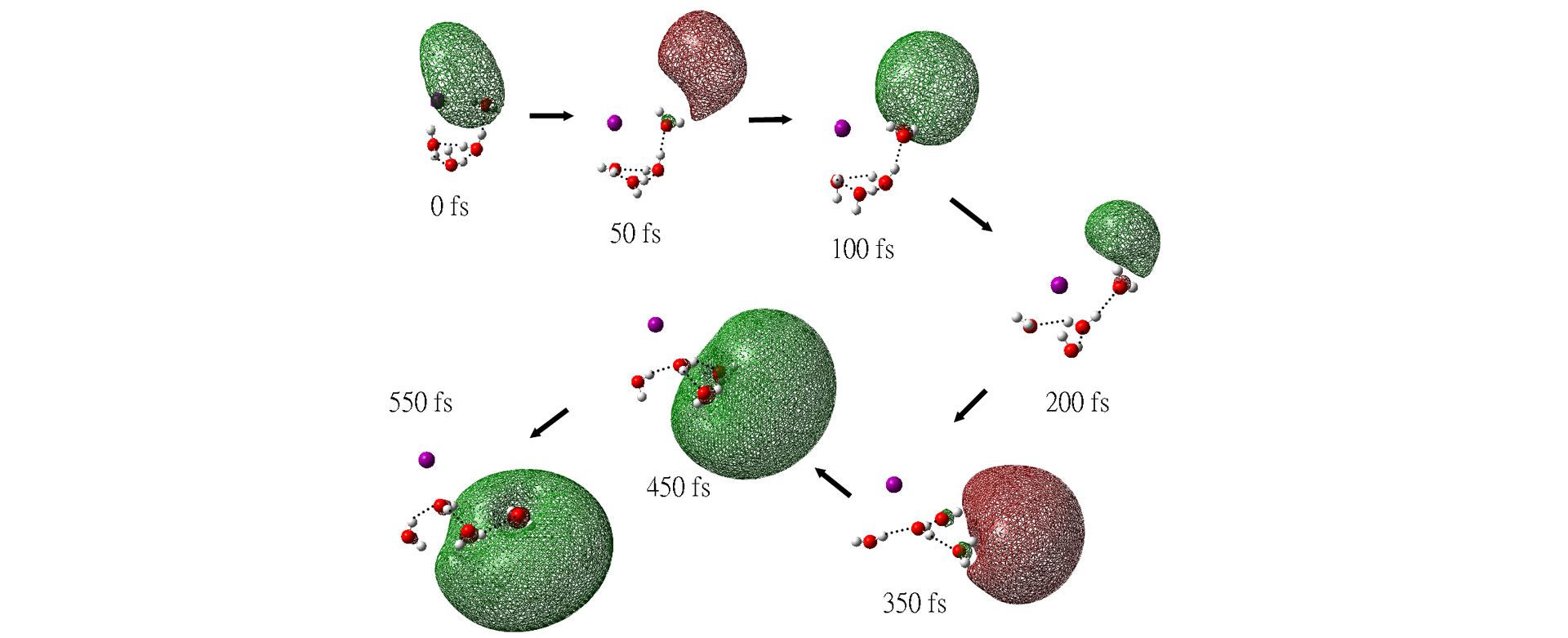
Effects of Iodine on the Relaxation Dynamics of a Photoexcited I-(H2O)4 Cluster
The Born-Oppenheimer molecular dynamics are used to examine the relaxation dynamics of the charge-transfer-to-solvent (CTTS) photoexcited electron in I-(H2O)4. The dynamics are initiated from the C
CO Oxidation on Ag(111) : the Catalytic Role of H2O
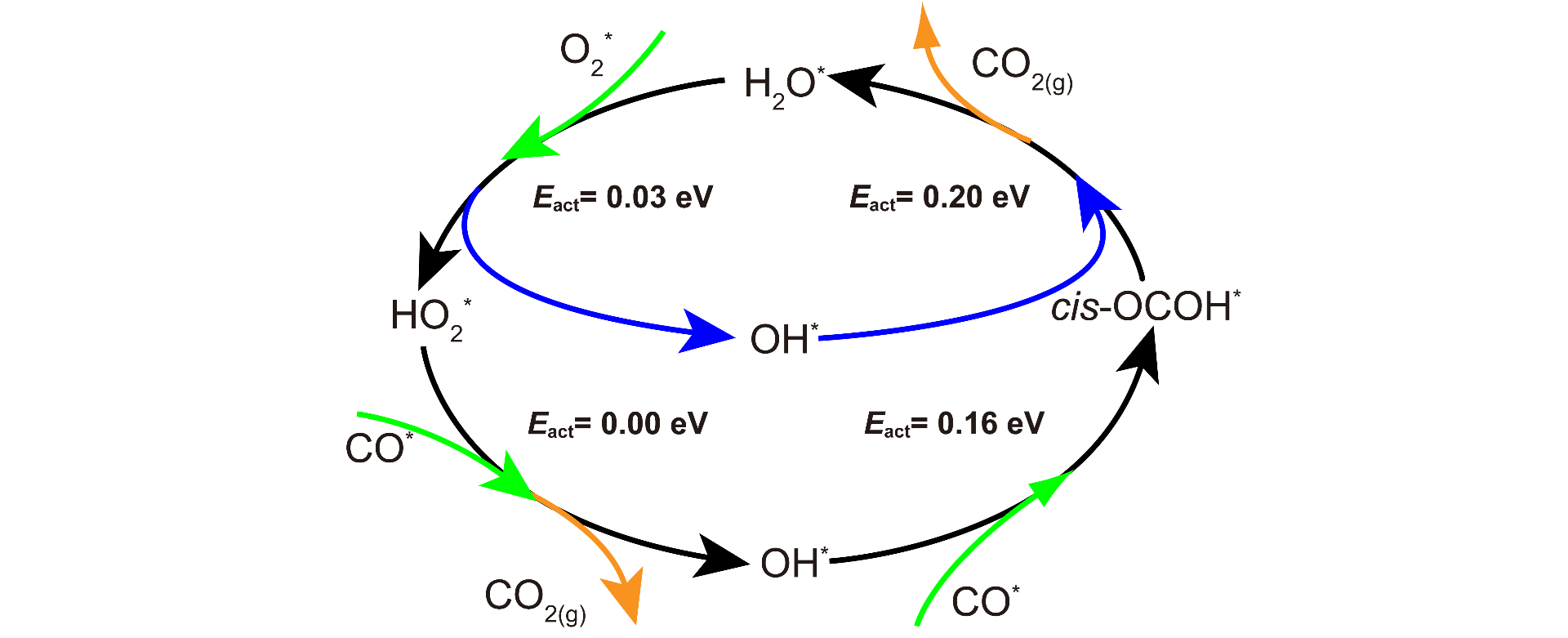
The reaction mechanism for the oxidation of CO on Ag(111) in the presence of trace amounts of water is investigated via density-functional-theory calculations. A four-step cycle for the reaction is proposed: (1) H2O + O2 → HO + HO2; (2) HO2 + CO → OH + CO2; (3) CO + OH → cis-OCOH; (4) cis-OCOH + OH → CO2 + H2O. In the mechanism, water is found to directly participate in the reaction as a catalyst, in addition to the previously proposed role of stabilizing the weakly adsorbed oxygen molecules on Ag(111). Moreover, HO2 is an important reaction intermediate, which is produced by transferring a hydrogen atom from water to an oxygen molecule. Because the overall reaction barrier is as low as 0.20 eV, the mechanism is expected to be operative at low temperatures.
Water-Gas-Shift Reaction on Reduced Gold-Substituted Ce1-x O2 (111) Surfaces: the Role of Au Charge
Density functional theory is employed to investigate the role of Au charge in the water-gas-shift (WGS) reaction on a CeO2(111) surface with a cerium atom replaced by a gold atom. The oxidation state of the gold atom, varied between +3 and -1, is controlled by altering the number and configuration of oxygen vacancies. The findings indicate that the Au3+ and Au− are not catalytically active for the WGS reaction because of a high energy barrier of +1.54 eV required to dissociate water and that of +1.40 eV to produce H2 and CO2, respectively. However, when the Au is in a modest oxidation state of +1, the overall reaction barrier for the WGS reaction via the carboxyl mechanism is reduced to be 0.79 eV ~ 0.98 eV. It therefore appears that Au species with an oxidation state of +1 play a significant role in the WGS reaction at low temperatures (T < ~550 K).
95 views